Unlocking Graphene Nanoelectronics: The Atomistic Dynamics of Area-Selective Hafnium Oxide Deposition
Research conducted by: Atiye Khosravi, Xichun Luo, Wenkun Xie, Xiaolei Zhang
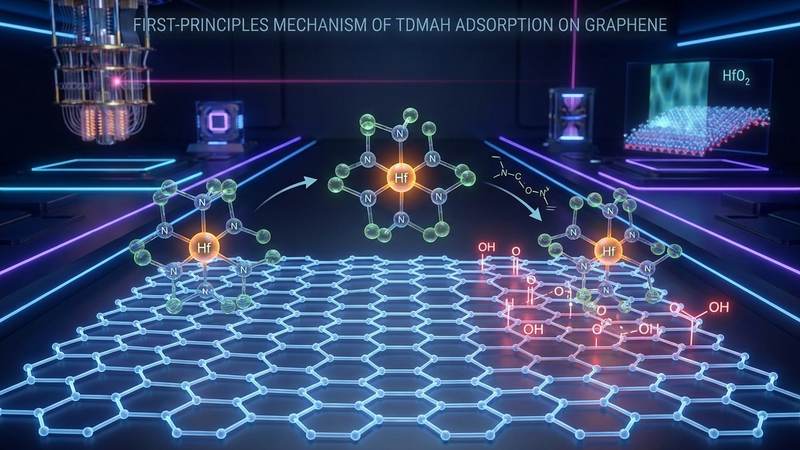
The groundbreaking study presented by these researchers provides an unprecedented atomistic framework for understanding the complex interfacial chemistry between advanced high-k dielectrics and graphene. By meticulously applying first-principles density functional theory, the team has successfully decoded the precise mechanisms governing the adsorption of tetrakis(dimethylamido)hafnium on both pristine and oxygen-functionalized graphene surfaces. Their work effectively bridges a critical knowledge gap in semiconductor manufacturing, detailing how two-photon laser oxidation can be utilized to overcome the inherent chemical inertness of graphene. This research not only clarifies the microscopic nucleation delays that have long plagued atomic layer deposition processes but also establishes a highly practical selectivity window for area-selective deposition. The dedication and analytical rigor demonstrated by Khosravi, Luo, Xie, and Zhang offer a transformative roadmap for the future of graphene-based field-effect transistors, quantum computing architectures, and next-generation nanoelectronics.
The Chemical Inertness of Pristine Graphene in Atomic Layer Deposition
Graphene has long been heralded as a miracle material within the realm of solid-state physics and materials science. Composed of a single layer of carbon atoms arranged in a two-dimensional honeycomb lattice, it boasts unparalleled electron mobility, exceptional mechanical strength, and remarkable thermal conductivity. These properties make it an ideal candidate for the channel material in next-generation field-effect transistors. However, the integration of graphene into commercial semiconductor devices faces a monumental hurdle: the deposition of high-quality gate dielectrics. Hafnium oxide is the industry standard high-k dielectric, utilized to prevent gate leakage currents while maintaining strong capacitive coupling. In traditional silicon manufacturing, hafnium oxide is deposited using atomic layer deposition, a technique that relies on sequential, self-limiting surface reactions to build highly conformal thin films one atomic layer at a time.
The fundamental issue with pristine graphene is its perfect sp2-hybridized carbon lattice. Unlike silicon, which features a native oxide layer rich in reactive dangling bonds and hydroxyl groups, pristine graphene is chemically inert. Its surface lacks the necessary active sites to initiate the chemisorption of the precursor molecules used in atomic layer deposition. When manufacturers attempt to deposit hafnium oxide onto pristine graphene using the standard precursor tetrakis(dimethylamido)hafnium, the molecules fail to anchor to the surface. Instead of forming a uniform, continuous film, the hafnium oxide grows in isolated, discontinuous islands. This island-growth mode, known as Volmer-Weber growth, results in severe pinhole defects, increased surface roughness, and catastrophic gate-leakage currents that entirely negate the performance benefits of the graphene channel. Understanding and overcoming this inertness without destroying the delicate electronic properties of the graphene lattice is one of the most pressing challenges in modern nanoelectronics.
Overcoming Inertness Through Two-Photon Laser Oxidation
To solve the nucleation dilemma, surface engineering techniques must be employed to artificially introduce reactive sites onto the graphene lattice. Traditional functionalization methods, such as wet chemical treatments, ozone exposure, or harsh plasma treatments, are often too aggressive. They tend to induce widespread structural damage, converting too much of the sp2 carbon network into sp3 hybridized defects, which drastically degrades the carrier mobility of the graphene. Enter two-photon laser oxidation, a highly sophisticated, non-destructive, and spatially precise technique that is revolutionizing surface functionalization.
Two-photon laser oxidation utilizes ultrafast femtosecond lasers operating at specific wavelengths where the photon energy is exactly half of the energy required to induce a chemical transition. Because the absorption of two photons must occur simultaneously to trigger the reaction, the oxidation process is highly non-linear and confined strictly to the focal point of the laser beam. This enables sub-diffraction-limit spatial resolution. By sweeping the laser across the graphene surface in a controlled pattern, researchers can selectively write highly localized regions of oxygen-containing functional groups while leaving the adjacent areas completely pristine.
The process predominantly introduces three types of oxygen functionalities to the carbon lattice: hydroxyl groups, carboxyl groups, and epoxide groups. These functional groups act as chemical anchors, eagerly awaiting interaction with the atomic layer deposition precursors. The beauty of two-photon laser oxidation lies in its ability to create a customized surface landscape. By tuning the laser power and exposure time, engineers can control the precise density and type of oxygen groups, effectively dialing in the exact level of surface reactivity required for optimal hafnium oxide nucleation, setting the stage for true area-selective atomic layer deposition.
First-Principles Density Functional Theory as an Atomistic Lens
To truly optimize the atomic layer deposition process on laser-functionalized graphene, empirical trial and error is insufficient. A deep, atomistic understanding of the precursor-surface interaction is required. This is where first-principles density functional theory becomes indispensable. Density functional theory is a quantum mechanical modeling method used in physics and chemistry to investigate the electronic structure of many-body systems. In this study, the researchers employed density functional theory to simulate the exact behavior of the tetrakis(dimethylamido)hafnium molecule as it approaches and interacts with various configurations of the graphene surface during the crucial first half-cycle of the atomic layer deposition process.
The computational models utilized extensive supercells to accurately represent the periodic boundary conditions of the two-dimensional material. The researchers calculated the adsorption energy, which is the exact amount of energy released or absorbed when the precursor molecule binds to the surface. A highly negative adsorption energy indicates a strong, stable, and spontaneous binding event characteristic of chemisorption. Conversely, a weak adsorption energy near zero suggests physisorption, where the molecule is only loosely held by weak Van der Waals forces. By systematically placing the precursor molecule over pristine graphene, as well as graphene functionalized with hydroxyl, carboxyl, and epoxide groups, the density functional theory simulations provided a rigorous, quantitative comparison of reactivity. This computational approach strips away macroscopic variables, allowing scientists to observe the fundamental quantum mechanical interactions that dictate thin-film growth at the sub-nanometer scale.
Tetrakis(dimethylamido)hafnium Adsorption Dynamics Explained
The simulations yielded profound insights into the adsorption dynamics of tetrakis(dimethylamido)hafnium. On pristine graphene, the interaction was predictably weak. The precursor molecule hovered above the perfectly smooth carbon lattice, held only by transient dipole interactions. The calculated adsorption energies were extremely low, confirming that pristine graphene essentially repels the precursor. In a real-world atomic layer deposition reactor, these weakly physisorbed molecules would be easily swept away during the inert gas purging phase of the cycle, explaining the severe nucleation delay and poor film quality observed in experiments.
However, the introduction of oxygen functional groups completely transformed the energy landscape. The study revealed that hydroxyl and carboxyl groups act as highly aggressive nucleation sites. When the precursor molecule approached a hydroxylated or carboxylated region, a profound chemical reaction occurred. The hafnium atom at the center of the precursor formed a strong, direct covalent bond with the oxygen atom of the functional group. This process often involved the breaking of the hafnium-nitrogen bonds within the precursor, leading to the dissociation of a dimethylamine ligand, which is the intended chemical pathway in an ideal atomic layer deposition half-cycle. The adsorption energies for these interactions were highly negative, indicating robust chemisorption.
Interestingly, the epoxide groups demonstrated an intermediate behavior. An epoxide consists of an oxygen atom bonded to two adjacent carbon atoms, forming a strained three-membered ring. While more reactive than pristine graphene, the epoxide groups did not facilitate binding as strongly as the hydroxyl or carboxyl groups. This intermediate binding state suggests that while epoxide-rich surfaces can support nucleation, they might require slightly higher thermal energy or longer precursor pulse times to achieve the same film density. These detailed adsorption dynamics confirm that the specific type of oxygen functional group is just as important as the overall density of functionalization when engineering graphene surfaces.
Decoding Charge Density Differences and Bader Charge Analyses
To validate the adsorption energies, the researchers delved deeper into the quantum state of the system by analyzing the charge density difference. A charge density difference map is a powerful visualization tool derived from density functional theory that illustrates exactly where electrons migrate during a chemical reaction. By subtracting the electron density of the isolated surface and the isolated precursor from the electron density of the combined, interacting system, researchers can pinpoint regions of electron accumulation and electron depletion.
In the case of pristine graphene, the charge density difference maps showed negligible electron redistribution, visually confirming the absence of chemical bonding. The electrons remained tightly bound to their respective original atoms. In stark contrast, the maps for the hydroxyl and carboxyl functionalized surfaces revealed massive electron rearrangement. A dense region of electron accumulation was observed directly between the hafnium atom of the precursor and the oxygen atom of the functional group, providing undeniable physical evidence of strong covalent bond formation. Concurrently, regions of electron depletion were observed around the hafnium-nitrogen bonds, confirming the weakening and breaking of the precursor's native ligands.
To quantify this visual data, the team utilized Bader charge analysis. Bader analysis partitions the continuous electron density of the system into distinct atomic volumes, allowing researchers to calculate the exact net charge on each individual atom before and after the reaction. The Bader charge data revealed a significant transfer of electron charge from the hafnium precursor to the functionalized graphene surface. The magnitude of this charge transfer correlated perfectly with the calculated adsorption energies. The hydroxyl and carboxyl groups induced the largest electron transfer, solidifying their status as the optimal nucleation sites for high-k dielectric growth.
Projected Density of States and the Shift Toward Strong Chemisorption
The final layer of computational verification came from the projected density of states. In solid-state physics, the density of states describes the number of different states at a particular energy level that electrons are allowed to occupy. By projecting this data onto specific atomic orbitals, researchers can determine exactly which electron orbitals are participating in the chemical bond. A strong chemical bond is typically characterized by the overlapping and hybridization of specific orbitals from the interacting atoms.
For the functionalized graphene systems, the projected density of states revealed a deep hybridization between the 5d orbitals of the hafnium atom and the 2p orbitals of the oxygen atoms on the functional groups. When the precursor bound to the hydroxyl or carboxyl groups, the energy peaks of the hafnium 5d and oxygen 2p orbitals aligned perfectly and broadened out, a classic signature of strong orbital mixing and covalent chemisorption. This hybridization lowered the overall energy of the system, creating a highly stable anchored state for the hafnium precursor.
Conversely, the projected density of states for the pristine graphene interaction showed sharp, localized peaks for both the precursor and the carbon lattice, with virtually no overlap or hybridization. The energy levels of the interacting orbitals remained distinct and unchanged, further corroborating the purely physisorptive nature of the interaction. By mapping out these orbital interactions, the researchers provided a complete, fundamental explanation for why laser oxidation so effectively shifts the surface chemistry from weak physisorption to robust chemisorption.
Area-Selective Atomic Layer Deposition and Future Nanoelectronic Applications
The most profound implication of this research is its application to area-selective atomic layer deposition. In traditional top-down semiconductor manufacturing, films are deposited globally across the entire wafer and then selectively removed using highly complex, expensive, and error-prone lithography and etching steps. Area-selective atomic layer deposition represents a paradigm shift toward bottom-up manufacturing, where films are engineered to grow only where they are desired, eliminating the need for destructive etching.
The atomistic mechanisms uncovered in this study define a precise selectivity window. Because pristine graphene exhibits near-zero reactivity and functionalized graphene exhibits exceptionally high reactivity, engineers can use two-photon laser oxidation to write intricate, nanoscale patterns of active sites onto a graphene wafer. When this patterned wafer is placed into an atomic layer deposition reactor, the hafnium oxide will rapidly nucleate and grow exclusively on the laser-oxidized patterns, while the pristine regions remain completely bare due to the severe nucleation delay.
This level of spatial control is critical for the development of advanced graphene field-effect transistors, where the gate dielectric must be perfectly aligned over the channel without encroaching on the source and drain contact regions. Furthermore, this bottom-up approach is vital for emerging quantum computing applications, where nanoscale dielectric barriers are required to isolate delicate quantum states. By providing a comprehensive first-principles understanding of tetrakis(dimethylamido)hafnium adsorption, this research bridges the gap between theoretical surface chemistry and practical semiconductor fabrication. It empowers engineers to optimize laser functionalization parameters and atomic layer deposition cycles, ensuring the flawless integration of high-k dielectrics and propelling graphene-based nanoelectronics closer to commercial reality.
Frequently Asked Questions
Q: What is the main challenge when depositing hafnium oxide on pristine graphene?
A: Pristine graphene has a perfectly bonded carbon lattice without any dangling bonds or reactive surface groups. This chemical inertness prevents the precursor molecules used in atomic layer deposition from anchoring to the surface, resulting in patchy, discontinuous films that cause severe electrical leakage in transistor applications.
Q: How does two-photon laser oxidation solve the nucleation problem?
A: Two-photon laser oxidation uses highly focused, ultrafast laser pulses to selectively introduce oxygen-containing functional groups onto the graphene surface. These groups act as chemical anchors that strongly attract and bind the atomic layer deposition precursors, enabling the growth of smooth, continuous dielectric films.
Q: What is first-principles density functional theory and why was it used here?
A: Density functional theory is a highly advanced quantum mechanical modeling technique used to calculate the electronic structure of molecules and materials. The researchers used it to simulate the exact atom-by-atom interactions between the precursor molecules and the graphene surface, eliminating guesswork and providing a fundamental understanding of the chemical bonding process.
Q: Which oxygen functional groups are most effective for precursor adsorption?
A: The computational simulations revealed that hydroxyl and carboxyl groups are the most effective. They facilitate strong covalent bonding and massive electron transfer with the hafnium precursor. Epoxide groups also improve reactivity compared to pristine graphene, but they offer intermediate binding strength rather than the robust chemisorption seen with hydroxyl and carboxyl groups.
Q: What is area-selective atomic layer deposition and why is it important?
A: Area-selective atomic layer deposition is a bottom-up manufacturing technique where materials are grown only in specific, pre-defined areas, eliminating the need for destructive etching processes. By selectively functionalizing graphene with a laser, manufacturers can force the hafnium oxide to grow only exactly where it is needed, which is crucial for building nanoscale transistors and quantum devices.
Conclusion
The integration of hafnium oxide high-k dielectrics onto graphene surfaces represents one of the most critical bottlenecks in the advancement of next-generation nanoelectronics. The comprehensive computational study conducted by Atiye Khosravi, Xichun Luo, Wenkun Xie, and Xiaolei Zhang provides a definitive, atomistic resolution to this challenge. By utilizing first-principles density functional theory, the research unequivocally demonstrates how pristine graphene's inherent inertness leads to weak physisorption, and how the strategic introduction of oxygen functional groups via two-photon laser oxidation radically alters the energy landscape to promote robust chemisorption. The detailed analyses of charge density differences, Bader charges, and projected density of states offer an unparalleled view into the quantum mechanics of thin-film nucleation. Ultimately, these findings establish a robust theoretical foundation for area-selective atomic layer deposition, offering semiconductor engineers the precise parameters required to fabricate flawless, high-performance graphene field-effect transistors and paving the way for the next era of high-speed, low-power computing.
Evaluate Our Quality
Serious about B2B integration? Test our premium Pulsed Electrical Resistive Carbon Heating turbostratic graphene in your lab. 100g sample packs available now.